
Manna, María Virginia. Médica residente de 2do año de Anatomía Patológica. Cátedra de Anatomía y Fisiología Patológicas, Facultad de Ciencias Médicas, Universidad Nacional de Rosario.
Saluzzi, Nadia. Médica patóloga. Cátedra de Anatomía y Fisiología Patológicas, Facultad de Ciencias Médicas, Universidad Nacional de Rosario.
CASO CLÍNICO
Paciente de cuatro años que consultó por dolor abdominal de cuatro días de evolución. Al examen físico, se observó a nivel tóraco-abdominal marcada diferencia de diámetro a nivel de la parrilla costal
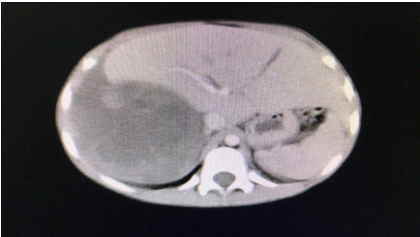
derecha y se palpó hepatomegalia a 4 cm por debajo del reborde costal. Se solicitó un laboratorio donde se constató velocidad de eritrosedimentación de 35 mm/hora (VN: hasta 10 mm/hora). La tomografía axial computarizada (TAC) revelaba a nivel renal derecho la existencia de una tumoración sólida, heterogénea, con áreas sugestivas de necrosis. La misma alcanzaba un tamaño de 120 x 114 mm y se hallaba en íntimo contacto con el parénquima hepático, sin plano neto de separación (Fig. 1). A nivel del tórax se destacaba la presencia de múltiples nódulos pulmonares en ambos campos, compatibles con secundarismo.
Como principales diagnósticos diferenciales principales se plantearon nefroblastoma versus hepatoblastoma, por lo cual se programó una punción biopsia guiada bajo TAC.
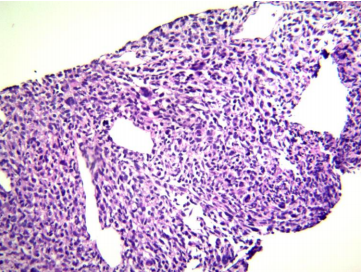
Los cilindros de punción se procesaron de manera habitual y el diagnóstico anatomopatológico fue de un NEFROBLASTOMA (tumor de Wilms) conformado predominantemente por componente blastemal (95%) y exiguo componente estromal (5%), con anaplasia focal (Fig. 2).
Clínicamente, y en correlación con las guías de diagnóstico y tratamiento propuesto por la Sociedad Internacional de Oncología Pediátrica, específicamente el Grupo de Estudio de Tumores Renales (SIOP-RTSG), se estadificó a la paciente como Estadio IV de su enfermedad. De acuerdo al protocolo se realizaron ciclos de quimioterapia, para posteriormente realizar el abordaje quirúrgico (1,2).
ANATOMÍA PATOLÓGICA
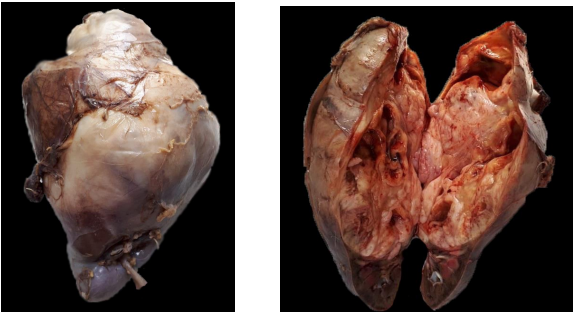
Fue remitida al servicio una pieza de nefrectomía total derecha de 24 x 15 x 10 cm y 1146 gr de peso. La superficie externa se hallaba tapizada por una cápsula lisa, brillante y translúcida. Al corte se identificó una lesión tumoral localizada en polo superior y nivel medio, de 16 x 11 x 10 cm.
Presentaba límites bien definidos, aspecto denso y color blanquecino, con focos amarillentos. En sectores se identificaron áreas de hemorragia y degeneración quística (Fig. 3). Se efectuó el procesamiento de acuerdo a las normas establecidas en el protocolo.
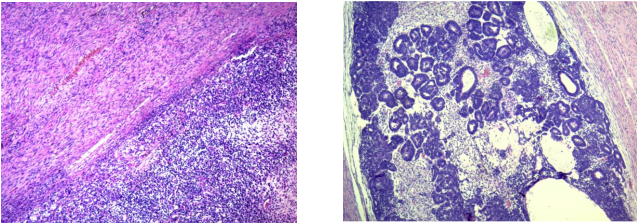
El diagnóstico anatomopatológico fue de un TUMOR DE WILMS DE TIPO MIXTO, representado por 70% del componente estromal, 20% blastemal y 10% epitelial (Fig. 4). La necrosis tumoral se estimó en un 20% y la anaplasia fue difusa. No se evidenció compromiso capsular, del seno renal ni imágenes de permeación vascular. Se observó además focos de diferenciación rabdomioblástica, islotes de cartílago y focos de osificación y calcificación.
El estudio inmunohistoquímico arrojó positividad para WT-1, desmina, miogenina y AE1/AE3 ( focal), S100 y CD57 negativos.
EVOLUCIÓN
Teniendo en cuenta el estudio histomorfológico, se concluyó que la paciente era portadora de un Tumor de Wilms de alto riesgo histológico y se evaluó la posibilidad de continuar nuevamente con ciclos de quimioterapia según protocolo ICE(5).
COMENTARIOS
El Nefroblastoma o Tumor de Wilms es una neoplasia embrionaria maligna que tiene su origen en el blastema nefrogénico. Corresponde al 90% de todos los tumores renales pediátricos malignos.
Generalmente afecta pacientes menores de 6 años y en el 5-10% pueden ser bilaterales ( sincrónicos o metacrónicos) y se asocian a alteraciones en el gen WT1 (11p13) y WT2 (11p15.5). Dicho tumor puede encontrarse vinculado a malformaciones renales y genitales; así como a síndrome de WilmsAniridia-Anomalía genital-Retraso mental, Síndrome de Beckwith-Wiedemann (onfalocelemacroglosia) y Síndrome de Denys-Drash (esclerosis mesangial difusa-pseudohermafroditismo 46 XY)(4).
El motivo de consulta más frecuente en estos casos es la detección de la masa abdominal sin ningún síntoma asociado. También se pueden manifestar con dolor abdominal, hematuria, hipertensión y anemia.
Macroscópicamente configuran masas renales esféricas grandes, lobuladas, rosado-grisáceas, con focos de aspecto necrótico y hemorrágico. Algunos tumores pueden presentar áreas friables, con sectores de degeneración quística o ser predominantemente quísticos. Para su procesamiento macroscópico es importante tener en cuenta que se debe mapear un corte completo de la lesión, e Identificar cada uno de los sectores (1) Histológicamente presenta tres componentes principales; blastemal, epitelial y estromal. Algunos tumores pueden presentar sólo uno o dos de los componentes mencionados(4).
A la hora del diagnóstico histopatológico, en aquellos pacientes que fueron estadificados y tratados según la SIOP-RTSG, se debe estimar el porcentaje de los cambios inducidos por la quimioterapia y
de los diferentes componentes del tumor; así como la presencia o ausencia de anaplasia (focal o difusa). Ésto permite la clasificación del riesgo histológico (alto, intermedio o bajo), pudiendo de esa Forma orientar la terapéutica posterior.
Si bien es posible arribar al diagnóstico por histomorfología, se recomienda complementar el estudio con técnicas de inmunohistoquímica. Los anticuerpos WT-1, PAX-8 y vimentina habitualmente son positivos en el componente blastemal; las citoqueratinas, EMA y CD56 en el epitelial; y vimentina, BCL2 y CD34 pueden positivizar en el componente estromal .
Entre los diagnósticos diferenciales a tener en cuenta vale mencionar al tumor rabdoide, neuroblastoma, sarcoma de Ewing y algunos carcinomas de células renales (6). Las características macroscópicas, microscópicas y el inmunofenotipo son de utilidad para diferenciar estas lesiones.
BIBLIOGRAFÍA
1- Vujanić, G.M., Gessler, M., Ooms, A.H.A.G. et al. The UMBRELLA SIOP–RTSG 2016 Wilms tumour pathology and molecular biology protocol. Nat Rev Urol 15, 693–701 (2018).
https://doi.org/10.1038/s41585-018-0100-3Vujanić, G.M., Gessler, M., Ooms, A.H.A.G. et al. The UMBRELLA SIOP–RTSG 2016 Wilms tumour pathology and molecular biology protocol. Nat Rev Urol 15, 693–701 (2018). https://doi.org/10.1038/s41585-018-0100-3
2-Bhatnagar, S., & Bhatnagar, S. (2009). Management of Wilms′ tumor: NWTS vs SIOP. Journal of Indian Association of Pediatric Surgeons, 14(1), 6. doi:10.4103/0971-9261.54811
3-Rosai J. “Ackerman´s Surgical Pathology.” Vol I 11th ed. Elsevier Mosby. 2019.
4-International Agency for Research on Cancer. WHO classification of tumours of the urinary system and male genital organs (IARC WHO classification of tumours) 4. Lyon: WHO/IARC Press; 2016.
5-Abu-Ghosh, A. M. (2002). Ifosfamide, carboplatin and etoposide in children with poor-risk relapsed Wilms’ tumor: a Children’s Cancer Group report. Annals of Oncology, 13(3), 460–469.
doi:10.1093/annonc/mdf028
6-Cajaiba, MM, & Reyes-Múgica, M.. (2007). Tumores renales de la infancia y adolescencia asociados a anomalías cromosómicas. Actas Urológicas Españolas, 31(9), 966-977.
gracias Virginia y Nadia por compartir este caso
Muy interesante y muy buena y concisa la descripcion y los comentarios. Las imagenes muy bien.
Saludos.
Valenti